DNA assembly tools used in the second generation of high-throughput sequencing technologies
Second-generation high-throughput sequencers have achieved cheaper and faster DNA sequencing methods, but they share a common shortcoming that reads are too short, ranging from tens of bp to hundreds of bp. Compared to the length of the chromosome of the creature, such a length of read troubles the next assembly work. The seemingly diverse biosphere actually uses only four nucleotides, A, T, G, and C. This means that there are repeating sequences in a very large number of fragments in a DNA sequence.
Therefore, it is sometimes difficult to judge the position of short reads in the original sequence during assembly. Reads A can be placed on multiple sites such as C, D, and E. Reads B seems to be placed on D, G, and H. This type of work can't be done by hand. You need to use a computer to assemble the readers according to certain rules and requirements.
The following describes some of the assembly software and features provided by non-equipment manufacturers.
1. Software name: Cross_match, Author: Phil Green, Brent Ewing and David Gordon
Website: http://
CROSS_MATCH is used for sequence alignment of proteins and nucleic acids using the Smith-Waterman-Gotoh algorithm. Improve operational efficiency through improvements to recursive relationships. Applicable to 1. Sort the reads according to the reference sequence. 2. Compare the contigs obtained by different methods. 3. Compare the sequence of contigs and cosmids after assembly.
2. Software name: Exonerate, Author: Guy S. Slater and Ewan Birney
Website: http://~guy/exonerate
The heuristic progressive algorithm is used to complete the sorting operation, which avoids the shortcomings of traversing and long-term, and also solves the problem that the heuristic progressive algorithm is difficult to implement.
3. Software name: MAQ, Author: Heng Li
Website: http://maq.sourceforge.net
MAQ quickly assembles reads according to a reference sequence, inferring various variants including SNPs, insertions, and deletions. In the sorting phase, the gapless arrangement is first searched for by the lowest mismatch score. In order to increase the speed, MAQ only uses up to 2 mismatched reads in the first 28 bp.
MAQ is designed with the need to handle human DNA sequences, and it can be easily run with low hardware requirements. In order to view the results, MAQ also provides an OpenGL-based browsing tool MAQview.
4. Software name: Mosaik, Author: Michael Strömberg and Gabor Marth
Website: http://bioinformatics.bc.edu/marthlab/Mosaik
MOSAIK can read a wide range of reads from tens of bp to hundreds of bp, according to the Smith-Waterman algorithm, according to the template to generate a gap arrangement. In addition, MOSAIK supports multi-threaded operation with up to 8 CPUs. It can fully support the three major serializers Roche 454, Illumina, AB SOLiD, and experimentally support Helicos.
5. Software name: BWA, Author: Li H. and Durbin R
Website: http://bio-bwa.sourceforge.net/
Burrows-Wheeler Aligner (BWA) allows the use of longer reference sequences and the alignment of relatively short nucleotide sequences. BWA performs two algorithms BWA-Short and BWA-SW. BWA-Short is suitable for reads shorter than 200 bp, the latter for lengths of approximately 100 kbp. Both can arrange a sequence with gaps. Although BWA-SW can work on shorter reads, its sensitivity is lower, and vice versa.
There are many types of DNA assembly tools, and several of them are typical. In addition, there are RMAP, SHRiMP, SOAP, SSAHA2, SXOligoSearch, etc., which are limited to the length of the article.
So how do you choose a software that suits your needs? There are several factors involved.
1. System running time, Figure 1 is the time consumption of several softwares for sorting different lengths of reads.

Figure 1 ( By Bala et al )
2. System hardware requirements, the following figure is a comparison of software consumption memory size.

Figure 2 (By Bala et al)
3. The difference in accuracy, the comparison results are shown in the figure below.

Figure 3 (By Bala et al)
4. Can you work on finding SNPs, SVs, etc.
5. Solexa, 454, SOLiD sequencing results output format is not the same, pay attention to the difference. Few software can support all formats.
The assembly of reads also involves another problem that has not been properly addressed so far. We break the long sequence of the genome because we don't know how the entire genome sequence is aligned and how to distinguish reads from different chromosomes. At the same time, the existing technology does not allow a complete genome sequence to be measured at one time. These short sequences must be assembled and restored to the original sequence (de novo assembly) using a computer-based high-speed computing capability according to certain algorithms.
Because there is no reference sequence, it is likely that the assembled sequence will be quite different from the original sequence. A sequence sequencing method called paired-end gives reads with a fixed spacing. as follows:
It is a very long human genome, so much longer than E.Coli's.
It i ***** ery *****huma*****ome,*****uch *****r th*****Coli ***
A clever way, but did not solve all the problems. Because the genome contains a large number of repeats, polymorphisms, sequencing errors, there are many possible pathways (graph theory), or loopbacks in a sequence. See below.
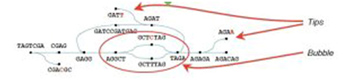
Some of the software that has achieved satisfactory results for these problems are as follows:
1. Software name Edena, Author: David Hernandez et al
Website: http://
2. Software name SHARCGS, Author: Juliane Dohm et al.
Website: http://sharcgs.molgen.mpg.de
3. Software name SSAKE, Author: René Warren et al.
Website: http://
4. Software name VCAKE, Author: William Jeck
Website: http://sourceforge.net/projects/vcake
5. Software name Velvet, Author: Daniel Zerbino and Ewan Birney
Website: http://
At present, the hope of finally solving the assembly problem is based on the advent of the third-generation high-throughput single-molecule sequencing technology. It is king to increase the length of each read. On the journey of life exploration, scientists have a long way to go.
SICHUAN UNIWELL BIOTECHNOLOGY CO.,LTD. , https://www.uniwellbio.com